BioComp - język do nauki symulacji modeli organizmów - dsl.biokomputery.pl
BioComp “Coded Life” to propozycja języka domenowo-specyficznego (DSL) do edukacji i wdrażania biocomputingu obejmuje elementy języka opisu eksperymentów, symulacji, designu i utrzymania systemów biokomputerowych. Motto “Coded Life” Łączy w sobie koncepcję kodowania (nawiązując do aspektu programistycznego) z życiem (odnosząc się do biologicznej strony projektu).
Składnia języka
BioComp jest zaimplementowany jako skrypt jezyka python z klasami dla modelu i funkcjami przetwarzania pliku YAML, generowania symulacji i grafu.
Plik biocomp.py modelu jest konwertowany do obiektow klas potrzebnyh do uruchamienia symulacji.
Plik YAML zawiera potrzebne informacje do wykonania symulacji:
- Definicje Molekularne
- Tworzenie Biologicznych Układów Logicznych
- Symulacje Eksperymentów
- Drukowanie i Inżynieria BioSystemów
- Monitorowanie i Utrzymanie Systemów
Zależności
pyparsing: Biblioteka do parsowania, potrzebna do przetwarzania DSL.matplotlib: Biblioteka do tworzenia wizualizacji, użyta do generowania wykresów wyników symulacji.numpy: Biblioteka do operacji na tablicach wielowymiarowych, używana do generowania danych do symulacji.graphviz: The addition of thegraphvizmodule is necessary for graph visualization.
Instalacja
Aby zainstalować te wymagania w swoim środowisku Python, wykonaj następujące kroki:
Stwórz i aktywuj wirtualne środowisko:
python -m venv env
source env/bin/activate # Na Windows użyj: env\Scripts\activate
lub If the user has Miniconda or Anaconda, I will provide instructions to create a new environment and install Graphviz within that environment:
conda create -n biokomputer python=3.12 graphviz
conda activate biokomputer
python main.py
Zainstaluj zależności z pliku requirements.txt:
pip install -r requirements.txt
pip install --upgrade pip
Symulacje bramek logicznych
Uruchomienie
Aby uruchomić tę konfigurację, wykonaj następujące kroki:
- Zapisz kod Python w pliku o nazwie
biocomp.py. - Zapisz zawartość pliku YAML w pliku o nazwie
biocomp.yaml. - Uruchom skrypt Python z pliku
biocomp.py, podając plik YAML jako argument:
python biocomp.py biocomp.yaml
Wzór Przetwarzania Danych Wejściowych:
\[ \text{output\_level} = \frac{\sin(t)}{2} + 0.5 \]
def run_simulation(simulation):
time = np.linspace(0, float(simulation.conditions["time"]), 100) # Generowanie 100 punktów czasowych
output_levels = np.sin(time) / 2 + 0.5 # Przykładowa funkcja poziomu wyjściowego
Symulacja bramki AND
molecules:
- type: Protein
name: Input1Prot
- type: Protein
name: Input2Prot
- type: Protein
name: OutputProt
logic_gates:
- gate_type: AND
input1: Input1Prot
input2: Input2Prot
output: OutputProt
biological_system:
name: BioCompSystem1
simulation:
conditions:
time: 100
temperature: 37
outputs:
- Protein OutputProt
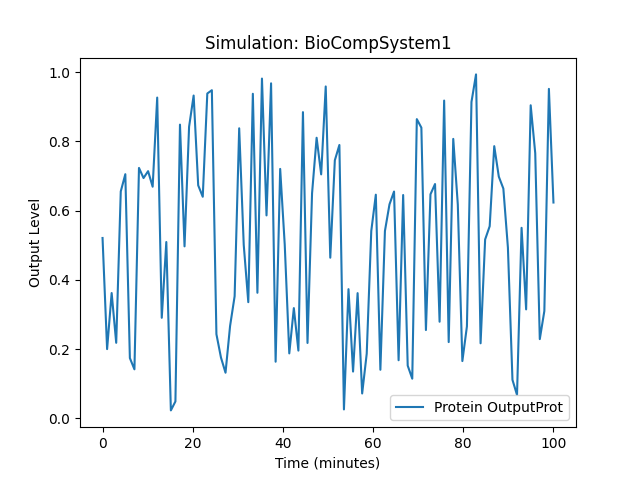
Interpretacja Wykresu
Wykresy generowane z symulacji opartej na pliku DSL pokazują dynamiczne zmiany w warunkach eksperymentalnych w funkcji czasu.
-
Oś X (czas): Przedstawia czas trwania symulacji w minutach. Parametr
simulation.conditions['time']ustala zakres czasowy symulacji. Cały przedział czasowy jest podzielony na 100 równych odstępów, dzięki czemu otrzymujemy szczegółowy wykres zmienności w czasie. -
Oś Y (poziom wyjściowy): Przedstawia poziom wyjściowy białka lub innej zmiennej wyjściowej, określonej przez
simulation.outputs. Funkcjanp.sin(time) / 2 + 0.5daje synusoidalne zmiany poziomu wyjściowego o amplitudzie 0.5 i przesunięciu pionowym o 0.5, co oznacza, że wartości zmieniają się w zakresie od 0 do 1. -
Linia wykresu: Jest to krzywa pokazująca zmiany poziomu wyjściowego w czasie. Nazwa białka lub zmiennej jest wyświetlana jako etykieta wykresu na podstawie zawartości
simulation.outputs[0].
Przykładowe Wyniki Wykresu
W przypadku symulacji opartej na przykładzie z wcześniejszym pliku 1/biocomp.yaml, uzyskany wykres może wyglądać tak:
- Symulacja: BioCompSystem1: Tytuł wykresu wskazuje, że symulacja jest wykonywana na systemie
BioCompSystem1. - Etykieta: Protein OutputProt: Etykieta na wykresie odnosi się do nazwy wyjściowego białka zdefiniowanego w symulacji.
Poniżej znajduje się przybliżona wizualizacja:
| ________
| / \
| / \________ x100 minutes
|_______/ \_______
0.0 100.0
Przykładowy Wykres ze Wskazaniem
- Wartości na osi Y zmieniają się synusoidalnie od poziomu 0 do 1.
- Wartości czasowe na osi X biegną od 0 do 100 minut.
Rzeczywiste Zastosowanie i Dane
W praktycznych zastosowaniach, dane wejściowe będą bardziej złożone i precyzyjne, bazujące na faktycznych pomiarach lub modelach biomolekularnych. Symulacje mogą prezentować poziomy ekspresji genów, aktywności enzymatycznej, stężeń cząsteczek sygnałowych i innych ważnych parametrów biologicznych w zależności od warunków eksperymentalnych.
Przykłady
Przykładowa sinusoidalna funkcja pokazuje podstawowe podejście do wizualizacji tych danych, ale rzeczywiste dane mogą być o wiele bardziej skomplikowane, zależnie od specyfiki symulacji i modelu biokomputerowego.
Te przykłady obejmują różne konfiguracje dla różnych typów biologicznych układów logicznych. Przykłady obejmują różne typy bramek logicznych, takie jak “AND”, “OR” i “NOT”. Jeśli w twojej klasie LogicGate zostały zaimplementowane poprawne działanie dla tych bramek, to te przykłady powinny dawać odpowiednie wyniki na podstawie ustawionych warunków w plikach YAML.
Jeśli nie zaimplementowano jeszcze działania dla tych bramek w klasie LogicGate, to należy to zrobić.
Aby przetestować twoją aplikację z tą funkcjonalnością, należy zapisać każdy plik YAML jako osobny plik.
BioCompSystem1
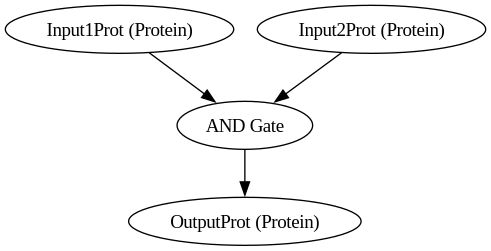
digraph {
Input1Prot [label="Input1Prot (Protein)"]
Input2Prot [label="Input2Prot (Protein)"]
OutputProt [label="OutputProt (Protein)"]
"AND Gate"
Input1Prot -> "AND Gate"
Input2Prot -> "AND Gate"
"AND Gate" -> OutputProt
}
python biocomp.py 1/biocomp.yaml
Running simulation for BioCompSystem1
Conditions: {'time': 100, 'temperature': 37}
Expected Outputs: ['Protein OutputProt']
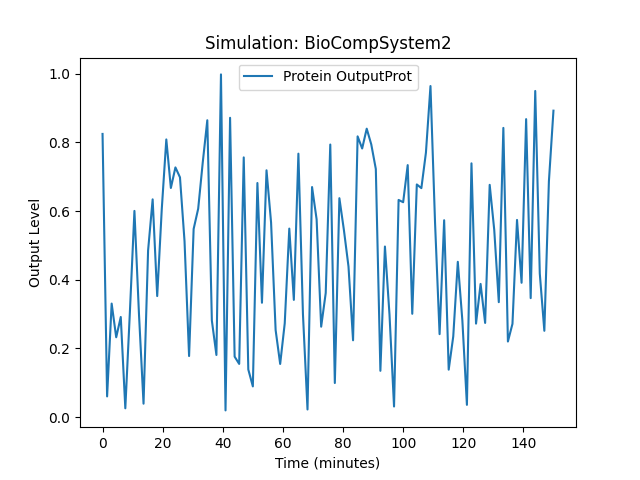
BioCompSystem2
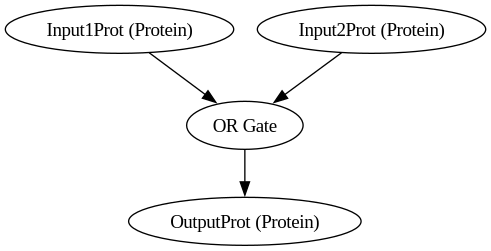
digraph {
Input1Prot [label="Input1Prot (Protein)"]
Input2Prot [label="Input2Prot (Protein)"]
OutputProt [label="OutputProt (Protein)"]
"OR Gate"
Input1Prot -> "OR Gate"
Input2Prot -> "OR Gate"
"OR Gate" -> OutputProt
}
python biocomp.py 2/biocomp.yaml
Running simulation for BioCompSystem2
Conditions: {'time': 150, 'temperature': 25}
Expected Outputs: ['Protein OutputProt']
BioCompSystem3
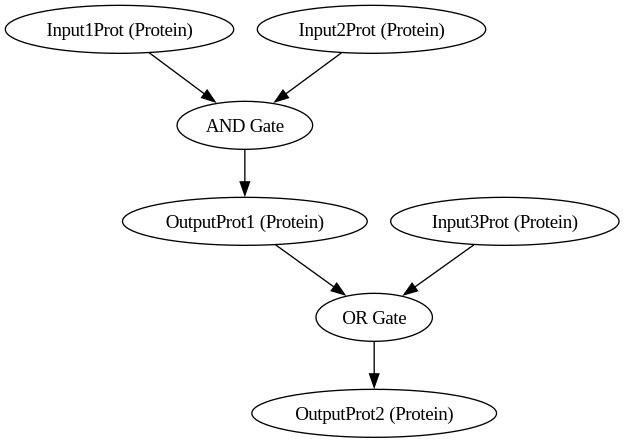
digraph {
Input1Prot [label="Input1Prot (Protein)"]
Input2Prot [label="Input2Prot (Protein)"]
Input3Prot [label="Input3Prot (Protein)"]
OutputProt1 [label="OutputProt1 (Protein)"]
OutputProt2 [label="OutputProt2 (Protein)"]
"AND Gate"
Input1Prot -> "AND Gate"
Input2Prot -> "AND Gate"
"AND Gate" -> OutputProt1
"OR Gate"
OutputProt1 -> "OR Gate"
Input3Prot -> "OR Gate"
"OR Gate" -> OutputProt2
}
python biocomp.py 3/biocomp.yaml
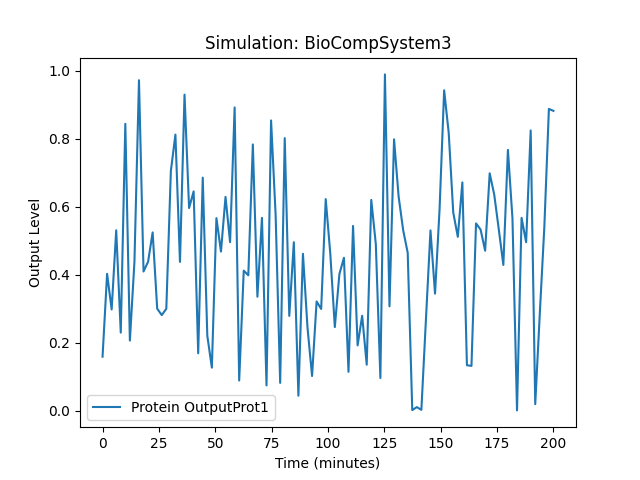
Running simulation for BioCompSystem3
Conditions: {'time': 200, 'temperature': 30}
Expected Outputs: ['Protein OutputProt1', 'Protein OutputProt2']
BioCompSystem4
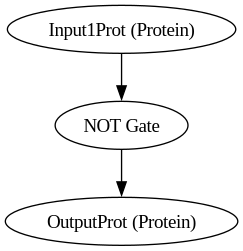
digraph {
Input1Prot [label="Input1Prot (Protein)"]
OutputProt [label="OutputProt (Protein)"]
"NOT Gate"
Input1Prot -> "NOT Gate"
"NOT Gate" -> OutputProt
}
python biocomp.py 4/biocomp.yaml
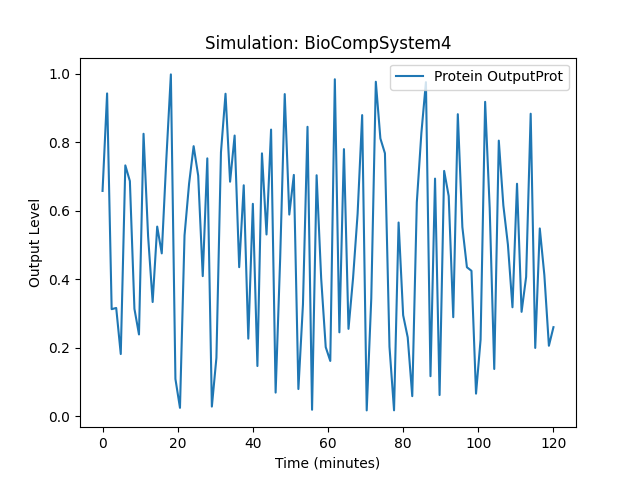
Running simulation for BioCompSystem4
Conditions: {'time': 120, 'temperature': 37}
Expected Outputs: ['Protein OutputProt']
Symulacje Grzybow
Aby przeprowadzić symulację, np. wzrostu grzybów, warto uwzględnić czynniki takie jak dostępność substancji odżywczych, temperatura, wilgotność i czas. Wzrost grzybów można modelować za pomocą różniczkowych równań wzrostu biologicznego, takich jak równanie Verhulsta (logistyczne równanie różniczkowe) lub modele podobne.
Wzór Symulacji Wzrostu Grzybów
Równanie logistyczne może być używane do modelowania wzrostu populacji organizmu, w tym grzybów:
\[ \frac{dN}{dt} = rN \left(1 - \frac{N}{K}\right) \]
Gdzie:
- ( N ) jest liczbą organizmów (np. masa biomasy grzybów),
- ( r ) jest wskaźnikiem wzrostu,
- ( K ) jest nośnością środowiska.
Skrypt fungi.py
Skrypt Pythona, który przetwarza dane wejściowe z plików YAML i wykonuje symulację wzrostu grzybów. Wzór logistyczny użyty w skrypcie pozwala modelować realistyczny wzrost populacji grzybów i może być dostosowany dzięki zmianie parametrów w plikach YAML. Skrypt wspiera przetwarzanie wielu plików YAML oraz opcjonalnie przetwarzanie wszystkich plików YAML w podanym folderze. Zawiera funkcje do generowania wykresów, tekstowych definicji grafów oraz graficznych reprezentacji grafów.
- Obsługa wielu plików YAML: Pobieranie wielu plików jako argumentów lub obsługa folderu zawierającego pliki YAML.
- Parsowanie: danych i tworzenie strukturę symulacji**,
- Symulacja: wzrostu grzybów za pomocą równania logistycznego,
- Wizualizacja wyników: Generowanie wykresów wzrostu biomasy.
- Generowanie tekstowej definicji grafu: Zapisywanie parametrów symulacji i wyników do pliku tekstowego.
- Graficzna reprezentacja grafu: Tworzenie grafów za pomocą Graphviz.
Wymagane biblioteki
Aby zapewnić pełną funkcjonalność, należy zainstalować następujące biblioteki:
yamlmatplotlibgraphviz
Można je zainstalować za pomocą pip:
pip install pyyaml matplotlib graphviz
FungiExperiment1
python fungi.py --folder ./11
digraph G {
node [shape=record];
"Simulation" [label="{Name: FungiExperiment1|Initial Population: 10|Growth Rate: 0.2|Carrying Capacity: 1000|Conditions: time=30, temperature=25, humidity=80|Outputs: Biomass}"];
}

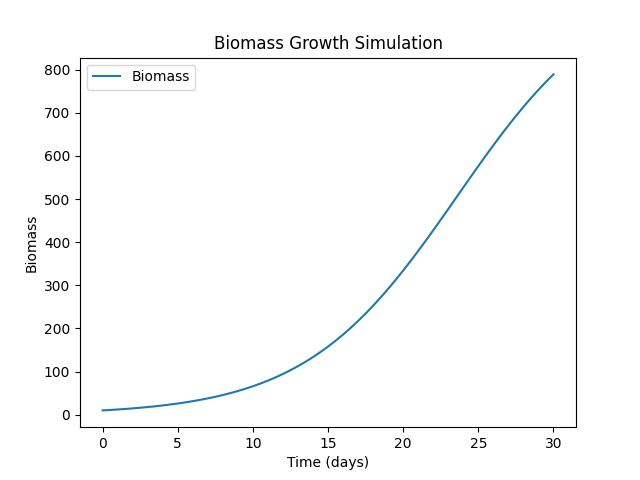
FungiExperiment2
python fungi.py --folder ./11
digraph G {
node [shape=record];
"Simulation" [label="{Name: FungiExperiment2|Initial Population: 5|Growth Rate: 0.1|Carrying Capacity: 500|Conditions: time=45, temperature=20, humidity=90|Outputs: Biomass}"];
}

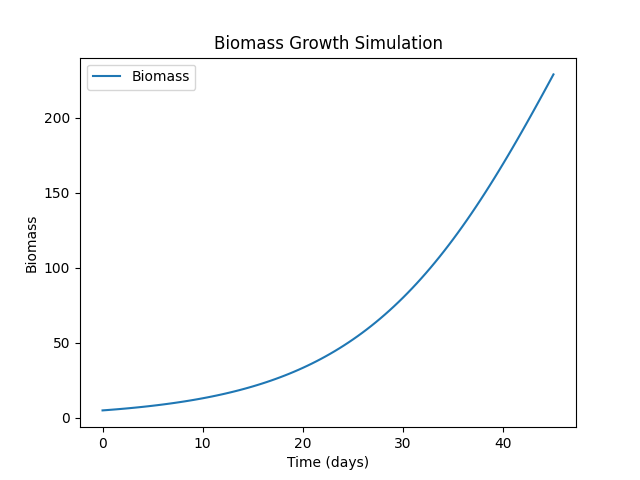
Symulacje Physarum
Aby zrealizować bardziej złożoną symulację wzrostu Physarum polycephalum (slime mold) jako hierarchicznej struktury komórkowej w kontekście modeli reakcyjno-dyfuzyjnych, należy przeanalizować kilka kluczowych procesów. Model reakcyjno-dyfuzyjny umożliwia symulację jak substancje (np. chemotaksyny) dyfundują oraz jak komórki reagują na te substancje, co prowadzi do wzrostu i formowania wzorca strukturalnego.
To jest dość zaawansowane zadanie, które można osiągnąć przy użyciu bibliotek takich jak NumPy do obliczeń oraz Matplotlib do wizualizacji. Do generowania grafów hierarchicznych użyjemy Graphviz.
Założenia:
- Hierarchiczna struktura: Model Physarum jako siatki komórkowej.
- Model reakcyjno-dyfuzyjny: Użyjemy prostego równania reakcyjno-dyfuzyjnego do modelowania rozprzestrzeniania się i reakcji chemotaksyn.
Równanie Reakcyjno-Dyfuzyjne
\[ \frac{\partial u}{\partial t} = D \nabla^2 u + f(u, v) \]
\[ \frac{\partial v}{\partial t} = D \nabla^2 v + g(u, v) \]
gdzie:
- ( u ) i ( v ) są koncentracją substancji chemicznych,
- ( D ) jest współczynnikiem dyfuzji,
- ( f(u, v) ) oraz ( g(u, v) ) są funkcjami reakcji.
Skrypt Python dla Modelu Hierarchicznej Struktury i Wykresu
Skrypt tworzy hierarchiczną strukturę Physarum jako maszynę reakcyjno-dyfuzyjną, przetwarza wiele plików YAML, a także generuje wizualizacje, tekstową definicję grafu i jego graficzną reprezentację. Wszystkie wygenerowane pliki są nazwane zgodnie z nazwami plików YAML, ale bez rozszerzenia .yaml.
Instalacja Wymaganych Bibliotek
Najpierw zainstaluj wymagane biblioteki:
pip install numpy matplotlib graphviz
PhysarumExperiment1
python physarum.py --folder ./21
- physarum.dot
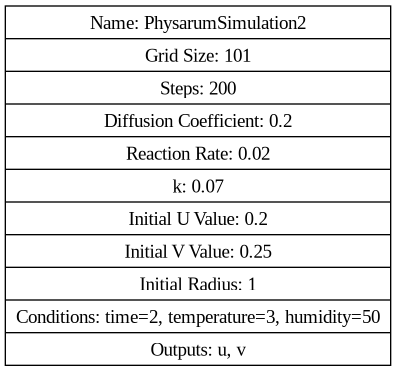
digraph G { node [shape=record]; "Simulation" [label="{ Name: PhysarumSimulation2 | Grid Size: 101 | Steps: 200 | Diffusion Coefficient: 0.2 | Reaction Rate: 0.02 | k: 0.07 | Initial U Value: 0.2 | Initial V Value: 0.25 | Initial Radius: 1 | Conditions: time=2, temperature=2, humidity=50 | Outputs: u, v }"]; }
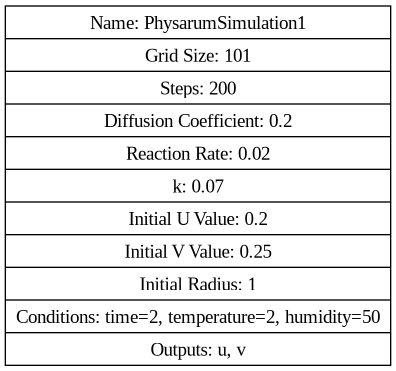
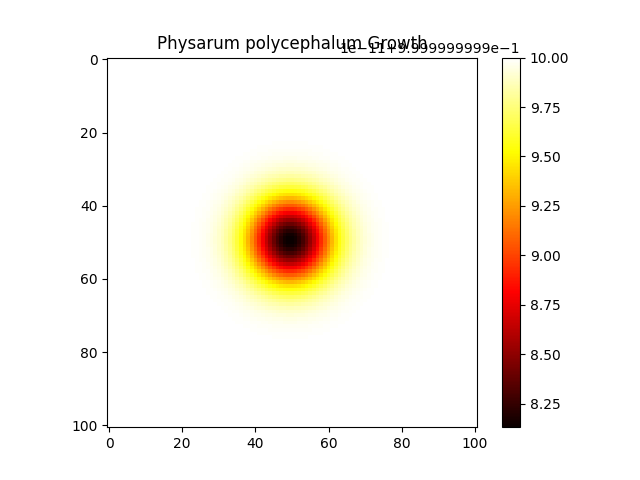
PhysarumExperiment2
python physarum.py --folder ./22
- physarum.dot
digraph G { node [shape=record]; "Simulation" [label="{ Name: PhysarumSimulation2 | Grid Size: 101 | Steps: 200 | Diffusion Coefficient: 0.2 | Reaction Rate: 0.02 | k: 0.07 | Initial U Value: 0.2 | Initial V Value: 0.25 | Initial Radius: 1 | Conditions: time=2, temperature=3, humidity=50 | Outputs: u, v }"]; }
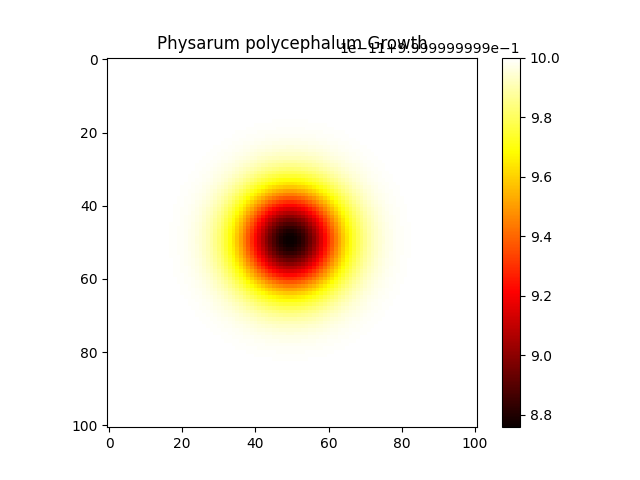
PhysarumExperiment3
python physarum.py --folder ./23
- physarum.dot
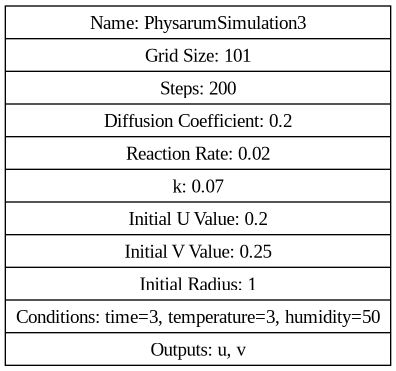
digraph G { node [shape=record]; "Simulation" [label="{ Name: PhysarumSimulation2 | Grid Size: 101 | Steps: 200 | Diffusion Coefficient: 0.2 | Reaction Rate: 0.02 | k: 0.07 | Initial U Value: 0.2 | Initial V Value: 0.25 | Initial Radius: 1 | Conditions: time=3, temperature=3, humidity=50 | Outputs: u, v }"]; }
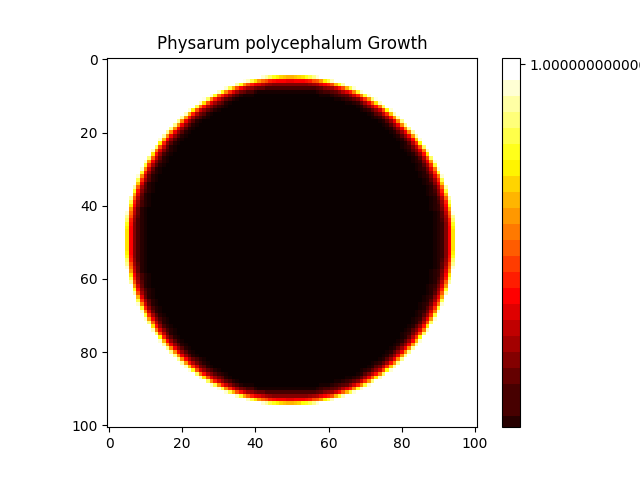
Star History
Contributions
CONTRIBUTION are always welcome:
- did you found an Issue or Mistake?
- do you want to improve the article?
- are you interested do join another git projects?
- have something to contribute or discuss? Open a pull request or create an issue.
Autor
Tom Sapletta
Na co dzień DevOps, ewangelista hipermodularyzacji, ostatnio entuzjasta biocomputing. Łączy doświadczenie w programowaniu i research-u poprzez wdrażanie nowatorskich rozwiązań. Szerokie spektrum zainteresowań, umiejętności analityczne i doświadczenie w branży owocują eksperymentalnymi projektami opensource.